
PBRER | Periodic Benefit Risk Evaluation Report – Simple Facts That You Never Know Before
Best Choices
Why PBRER?
It can be hard to keep track of all the new information about a medicinal product and how it impacts the balance of benefits and risks.
With so much new information coming out all the time, it can be hard to know if a product still has a good balance of benefits and risks.
The Periodic Benefit-Risk Evaluation Report (PBRER) is an essential report for evaluating the balance of benefits and risks of a medicinal product. The PBRER helps you stay up-to-date on new information, so you can make informed decisions about medical products.
What is a Periodic Benefit Risk Evaluation Report (PBRER)?
A Periodic Benefit Risk Evaluation Report (PBRER) is a comprehensive safety update report that is produced as part of the safety monitoring process for any medicinal product undergoing clinical trials or post-market safety analysis.
This report documents adverse events or safety issues that have arisen over the course of the trial or analysis, including information about patient enrollment and demographics and data on exposure to the drug, efficacy results, and possible risk factors.
By compiling all this information into a single document, the PBRER helps to ensure that safety considerations are always prioritized throughout each step of the development and regulatory decision process.
Ultimately, this approach allows researchers and healthcare professionals to maintain a clear view of any potential risks associated with a given medicinal product, allowing them to make well-informed decisions about its use.
Final word
The purpose of PBRER is to provide an evaluation of whether or not the benefits outweigh any risks. It takes into account safety, efficacy, and effectiveness throughout its lifecycle so that you can make informed decisions about your products’ fate for your patient.
What are PBRER Objectives?
When a new drug or device is approved by the FDA for circulation in America it’s not just enough to show that they’re safe and effective.
In order words you have your work cut out for yourself trying to get into one of these clinical trials since there are only so many spots available!
The need for continuing analysis of relevant safety, efficacy, and effectiveness information throughout the lifecycle is a priority in clinical practice.
The patient population studied may be more diverse than those included within trials (including older adults with multiple co-morbid conditions) as well an event too rare to occur by chance such severe liver injury can happen at any time during our lives!
The PSUR and PBRER provide a comprehensive picture of the safety profile for approved medicinal products.
The risk assessment is most meaningful when considered in light of its benefits, so it’s no surprise that this newer report places more emphasis on them than before- especially since there can sometimes be changes due to new information becoming available or studies being published over time which could affect how we perceive certain risks associated with using one particular medication regime (or even none).
Final word:
A PBRER’s primary goal is to provide a succinct, thorough, and critical examination of new or emerging data on a drug’s risks and benefits relative to its approved indications in order to assess the drug’s overall benefit-risk profile.
In the context of cumulative information, the PBRER should include an assessment of new information about the medicinal product that was made available to the MAH during the reporting interval. Therefore, providing cumulative and interval safety data information.
What is the Role of PBRER in clinical trials?
What are PBRER reports about?
PBRER reports are:
- Describing relevant new safety facts that may affect the pharmaceutical product’s benefit-risk profile.
- Describe any significant new efficacy or effectiveness data that have emerged throughout the reporting period.
- Determining whether the data the MAH collected during the reporting period is consistent with knowledge of the medicinal product’s benefits and risks from prior experience.
- Recognizing crucial recent safety findings for an integrated benefit-risk evaluation for approved indications
What are PBRER Sections consist of?
PBRER reports contain:
I. Introduction
1- IBD
2- reporting interval
3- medicinal product(s) –
- mothed of action,
- therapeutic class(es),
- dose(s),
- route(s) of administration,
- formulation(s)
4- a brief description of the approved indication(s) and population(s);
5- a brief description and explanation of any information that has not been included in the PBRER
6- the rationale for submission of multiple PBRERs for the medicinal product, if applicable
II- Marketing approval status
PBRER should provide
- a brief narrative overview including the date of first approval,
- indication(s),
- approved dose(s),
- and were approved
III- Actions are taken in the reporting interval for safety reasons
The PBRER should include a description of significant safety-related actions that have been taken during the reporting interval, either by an investigational use or marketing experience.
This can be anything from
- how many adverse events were reported to what kind they were (heart attacks)?
The MAH/ Sponsor may also need input into certain decisions such as whether participant withdrawals would affect their trial results; if so then this information would go through both monitoring committees AND ethics panels before being made final.
- A significant influence on the benefit-risk profile of the approved medicinal product; and/or
- An impact on the conduct of a specific clinical trial(s) or on the overall clinical development program.
Relevant updates to previous actions should also be summarised in this section. Examples of significant actions taken for safety reasons include:
- Refusal to authorize a clinical trial for ethical or safety reasons;
- Complete clinical trial suspension or early termination of an ongoing clinical trial because of safety findings or lack of efficacy;
• Recall of investigational drug or comparator;
• Failure to obtain marketing approval for a tested indication, including voluntary withdrawal of a
marketing application;
• Risk management activities.
IV – Changes to reference safety information
The PBRER should list any significant changes to the reference safety information within each reporting interval.
Such changes might include information relating to contraindications, warnings precautions for adverse drug reactions (ADRs)and interactions; important findings from ongoing clinical trials.
V- Estimated exposure and use patterns
The PBRER should provide estimates of the size and nature
of the population exposed to medicinal products.
Section 5 provides information on cumulative exposure in clinical trials, while section 6 offers calculations for estimated patient doses based on reported data from market release periods.
If there’s been any change in methodology then both approaches will be detailed so you can stay updated with your knowledge gap!
VI- Cumulative and interval patient exposure from marketing experience
1- Cumulative numbers of subjects from ongoing and completed clinical trials.
2- Important differences among trials in dose, routes of administration, or patient populations can be noted in the tables.
3- If clinical trials have been or are being performed in special populations.
4- When there are substantial differences in duration of exposure between subjects randomized to the investigational medicinal product or comparator(s), or disparities in the duration of exposure between clinical trials, it can be useful to express exposure in subject-time (subject-days, -months, or – years).
VII- Cumulative and interval patient exposure from marketing experience
The data should be presented according to the following categories:
1. Post-approval (non-clinical trial) exposure
2. Post-approval use in special populations
3. Other post-approval use
PBRER template
Here is an assessment of the risk by periodic benefit-risk evaluation reports of any medical product for safety update reports in post-marketing surveillance.
PBRER table of contents
Table of Contents
1. Introduction
2. Worldwide Marketing Approval Status
3. Actions Taken in the Reporting Interval for Safety Reasons
4. Changes to Reference Safety Information
5. Estimated Exposure and Use Patterns
5.1 Cumulative Subject Exposure in Clinical Trials
5.2 Cumulative and Interval Patient Exposure from Marketing Experience
6. Data in Summary Tabulations
6.1 Reference Information
ICH guideline E2C (R2) on periodic benefit-risk evaluation report (PBRER)
EMA/CHMP/ICH/544553/1998 Page 13/45
6.2 Cumulative Summary Tabulations of Serious Adverse Events from Clinical Trials
6.3 Cumulative and Interval Summary Tabulations from Post-Marketing Data Sources
7. Summaries of Significant Findings from Clinical Trials during the Reporting Period
7.1 Completed Clinical Trials
7.2 Ongoing Clinical Trials
7.3 Long-Term Follow-up
7.4 Other Therapeutic Use of Medicinal Product
7.5 New Safety Data Related to Fixed Combination Therapies
8. Findings from Non-Interventional Studies
9. Information from Other Clinical Trials and Sources
10. Non-Clinical Data
11. Literature
12. Other Periodic Reports
13. Lack of Efficacy in Controlled Clinical Trials
14. Late-Breaking Information
15. Overview of Signals: New, Ongoing, or Closed
16. Signal and Risk Evaluation
16.1 Summary of Safety Concerns
16.2 Signal Evaluation
16.3 Evaluation of Risks and New Information
16.4 Characterisation of Risks
16.5 Effectiveness of Risk Minimisation (if applicable)
17. Benefit Evaluation
17.1 Important Baseline Efficacy/Effectiveness Information
17.2 Newly Identified information on Efficacy/Effectiveness
17.3 Characterisation of Benefits
18. Integrated Benefit-Risk Analysis for Approved Indications
18.1 Benefit-Risk Context – Medical Need and Important Alternatives
18.2 Benefit-Risk Analysis Evaluation
19. Conclusions and Actions
20. Appendices
PBRER reporting timelines
Before talking about PBRER reporting timelines, let’s know important terms,
What is IBD?
IBD is the date of the first marketing approval for any product containing the active substance granted to any company in any country in the world
What is a data lock point?
The DLP is the date designated as the cut-off for data to be included in a PBRER.
What if there are still going clinical trials?
In the future, it will be possible to have a single report that combines data from both DSUR and PBRER ( periodic benefit-risk evaluation report).
This approach has two major benefits: firstly by harmonizing their content they can share certain sections/modules; secondly, since there is only one annual submission this saves time during processing as well as makes life easier for sponsors who must prepare these documents at different times because of how long ago some events happened (such influenza outbreaks).

PBRER frequency
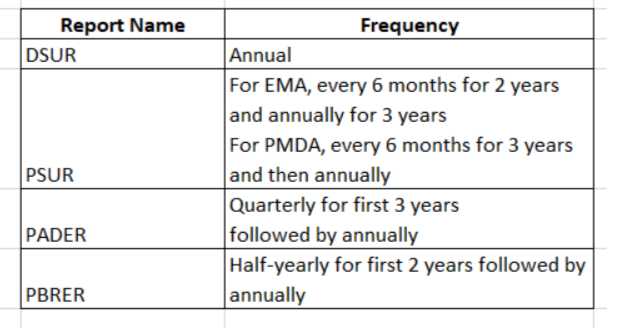
The need for submission of periodic benefit-risk evaluation reports and frequency depends on various factors such as approval dates, length of product that has been available, etc., but it is subject to national or regional regulatory requirements.
The PBRER format and content were developed to help companies submit reports that cover periods of 6 months or more. Once a drug has been marketed for several years, the frequency in which these documents must be submitted may change depending on whether you’re located within North America vs another region; however, it is still important to follow all guidelines relevant to your specific situation.

What are Ad hoc PBRERs?
Ad hoc PBRERs are reports outside the routine reporting requirements and may be requested by some regulatory authorities. Where an ad hoc report is requested and a PBRER has not been prepared for a number of years, it is likely that a completely new report will need to be prepared by the MAH.
What is the time interval between the data lock point and the PBRERs submission?
- periodic benefit risk evaluation report is covering intervals of 6 or 12 months: within 70 calendar days;
- periodic benefit risk evaluation report is covering intervals in excess of 12 months: within 90 calendar days;
- ad hoc periodic benefit risk evaluation report: 90 calendar days unless otherwise specified in the ad hoc request.
PBRER signals and riskes map
Risk evaluation report pbrer map
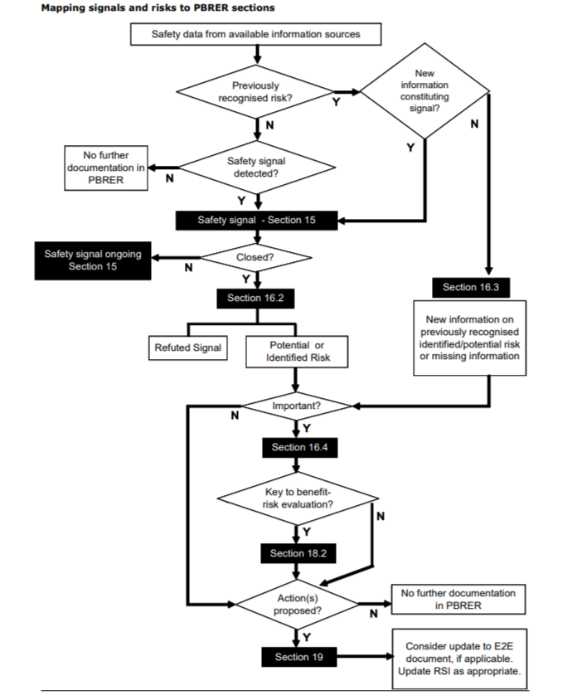
What is the role of PBRER in pharmacovigilance?
After the marketing authorization for a medicinal product has been granted, manufacturers are required to prepare and submit periodic benefit risk evaluation reports (PBRERS). These pharmacovigilance documents provide an in-depth look at how well patients can balance their risks versus rewards when taking their medications.
ICH E2C(R2) PBRER guidance
ICH is a regulatory authority in the EU and is responsible for drug efficacy and effectiveness by monitoring the safety information of medicinal products for the assessment of the risk to update the benefit-risk profile of medicinal products.
ICH E2C(R2) is a guidance document that provides recommendations for creating benefit-risk profiles for pharmaceutical products.
This guidance focuses on understanding the safety information associated with a drug, including both the potential adverse effects and any risks associated with its administration.
By taking these factors into account, companies can develop strategies to manage potential risks and ensure that their products provide maximum benefit to patients.
Overall, ICH E2C(R2) provides important insight into how to create effective benefit-risk profiles during drug administration, helping to ensure that drugs are safe and effective for use in clinical practice.
GVP & Pbrer
1. Pharmacovigilance is the science and activities relating to the detection, assessment, understanding, and prevention of adverse effects or any other drug-related problems.
2. Good pharmacovigilance practice (GVP) is a set of principles and guidelines that provide a framework for the conduct of pharmacovigilance activities.
3. The purpose of GVP is to ensure that pharmacovigilance activities are conducted in a manner that protects public health and safety.
4. GVP is relevant to all phases of a medicinal product’s life cycle, from pre-marketing to post-marketing.
5. PBRERs are post-authorization safety studies that are conducted to assess the risk-benefit balance of a medicinal product after it has been authorized for use in humans.
PBRER VS PSUR
1. PBRER
PBRER stands for Periodic Benefit-Risk Evaluation Report. It is a type of report that is submitted to health authorities on a periodic basis, typically annually. PBRERs contain detailed information on the benefits and risks of a medicinal product, as well as any new information that has emerged since the last report was submitted.
2. PSUR ( periodic safety update reports )
PSUR stands for Post-Authorization periodic safety update reports. It is a type of report that is submitted to health authorities on a periodic basis, typically every 6 months. PSURs contain detailed information on the safety of a medicinal product, as well as any new information that has emerged since the last report was submitted.
3. Difference
The main difference between PBRERs and PSURs( periodic safety update reports ) is the type of information that they contain. PBRERs focus on both the benefits and risks of a medicinal product, while PSURs only focus on safety. Additionally, PBRERs are typically submitted annually, while PSURs are typically submitted every 6 months.
4. Purpose
Both PBRERs and PSURs are used by health authorities to assess the safety and efficacy of medicinal products. These reports help to ensure that medicines are safe and effective for patients and that any new information about the medicine is promptly shared with authorities.
5. Reporting Requirements
PBRERs and PSURs are typically required for all medicines that are authorized for use in humans. However, there may be some exceptions for certain types of medicines, such as those that are used in clinical trials or those that have been authorized for use in emergency situations
PBRER VS DSUR
1. PBRER
PBRER stands for Periodic Benefit-Risk Evaluation Report. It is a type of report that is prepared by pharmaceutical companies to assess the benefits and risks of a medication on a regular basis. The reports are submitted to regulatory agencies, such as the FDA, and are used to help make decisions about whether or not medication should remain on the market.
2. DSUR
DSUR stands for Development Safety Update Report. It is a type of report that is prepared by pharmaceutical companies to assess the safety of medication during its development process. The reports are submitted to regulatory agencies, such as the FDA, and are used to help make decisions about whether or not medication should be approved for use.
3. Differences
There are several key differences between PBRERs and DSURs. First, PBRERs are prepared on a periodic basis (usually annually), while DSURs are only prepared when there is new safety information to report. Second, PBRERs focus on both the benefits and risks of a medication, while DSURs only focus on safety. Finally, PBRERs are used to make decisions about whether or not medication should remain on the market, while DSURs are used to make decisions about whether or not medication should be approved for use.
PADER VS PBRER
1. PBRER
PBRER stands for Post-Marketing Review of Experimental Results. It is a type of scientific study that is conducted after a new drug or medical device has been approved for use by the FDA. The purpose of a PBRER is to gather additional information about the safety and efficacy of the product in order to ensure that it is safe and effective for use by the general public.
2. PADER
PADER stands for Post-Approval Data Evaluation and Review. It is a type of scientific study that is conducted after a new drug or medical device has been approved for use by the FDA. The purpose of a PADER is to gather additional information about the safety and efficacy of the product in order to ensure that it is safe and effective for use by the general public.
3. Difference between PBRER and PADER
The main difference between PBRER and PADER is that PBRER is conducted after a new drug or medical device has been approved for use by the FDA, while PADER is conducted after a new drug or medical device has been approved for use by the FDA. Additionally, PBRER studies are typically larger and more comprehensive than PADER studies.
General Pharmacovigilance Terms You Should Know
What is the cumulative reporting interval in pbrer?
The cumulative reporting interval in pbrer is the time period within which any adverse drug reactions that occur must be reported to regulatory authorities.
This reporting interval is typically set by regulatory authorities and is determined based on a variety of factors, including the type and severity of the drug being administered, as well as the frequency with which it is used.
Generally speaking, reporting intervals can vary greatly depending on how widely a drug is used and how serious its side effects are.
For instance, less common and more severe drugs may require reporting at shorter or more frequent intervals than more commonly used drugs with milder side effects.
Regardless of these individual reporting times, however, all reports must be carefully tracked over time in order to ensure the accuracy of information provided to regulatory authorities.
Ultimately, this process helps to keep drug administration safe and effective for patients across the globe.
What is the difference between safety efficacy and effectiveness?
There are a few key differences between safety efficacy and effectiveness as it relates to drug administration.
At the most fundamental level, safety efficacy refers to the drug’s potential for harm or injury, while effectiveness refers to the drug’s ability to produce the intended benefits or effects.
Moreover, drug safety is determined by regulatory authorities such as the FDA based on data from clinical trials assessing both short-term and long-term risks, whereas drug efficacy is primarily evaluated based on the results of animal trials prior to drug marketing.
Finally, safety and efficacy must both be considered when determining the risk-benefit profile of a medicinal drug.
While many factors influence drug effectiveness, safety is always critically important in preventing adverse drug reactions and protecting patients against risk.
Overall, these distinctions underscore the complex nature of drug research and development and highlight why medications must be rigorously tested before they are made available to consumers.
What is the importance of regulatory authorities?
Regulatory authorities are an essential part of the healthcare system. These regulatory bodies play a key role in ensuring that the medications and treatments we use are both safe and effective.
They act as guardians of public health, monitoring the risks associated with medicines and other healthcare interventions, and they work to protect the well-being of patients. Furthermore, regulatory authorities help to facilitate scientific advances by conducting clinical trials and evaluating new treatment options, so that we can continue to provide effective care for our patients.
Overall, regulatory bodies are critical to ensuring that we have access to quality healthcare while minimizing any potential risk or harm to patients. Without regulatory authorities, we would be much more vulnerable to any dangers that may lurk within our medications or treatments. Therefore, their importance is clear, both for individual healthcare professionals and for the wider society at large.
What does it take to get approved medicinal products?
Getting approved for medicinal products can be a long and complicated process.
First of all, companies must have extensive testing data that supports their proposed product.
This testing may require several years to complete, so the companies must start working on their product very early on. Along with robust scientific evidence, these companies must also submit extensive documentation about their products, including information about potential side effects, drug interactions, and dosage recommendations.
Finally, companies must often conduct trials under strict guidelines to ensure that their products are safe and effective for patients. Overall, it takes a combination of scientific knowledge and careful planning to get approved and approved medicinal products onto the market.
FAQs
What is pbrer meaning?
It is a periodic benefit-risk evaluation report.
Where to find pbrer in GVP modules?
In E2C (R2)
Reference
E2C (R2) Step 5 Periodic benefit-risk evaluation report (PBRER)
Guideline on Periodic Benefit Risk Evaluation Report (PBRER)
Conclusion
By now you have found the importance of PBRER, if you want to know more about all types of reports is pharmacovigilance in the simplest way, click here.




